Throughout the remainder of this presentation, when showing parental contributions to a cross or mating, and the statistical outcome, I will present all the parental genes or the genotype for the trait being discussed. When discussing a gene that has a mutant allele, its non-mutant allele is referred to as the wild-type or natural allele. The wild-type trait is the most common phenotype in natural populations, whose breeding is not controlled by humans. The accepted method for symbolizing genes is capitals for dominant alleles, small letters for recessive alleles and “+” (or mutant allele with superscript “+” following) for the wild-type or natural allele. For example, the pied trait, which is a recessive mutation, would be symbolized as “pd”. For the wild-type allele or non-pied gene, this would be symbolized as “+” or “pd+”. Note that in the latter case although the wild-type is a dominant gene it is not capitalized. When expressing the wild-type you use the same letter case as the mutation gene followed by a superscript “+” sign.
Capital X and capital Y will only be used to represent the sex chromosomes. (Note: Use of X and Y to represent avian sex chromosomes is a deviation from normal genetic doctrine, which uses Z and W.) I will symbolize sex-linked genes as the sex chromosome with the gene symbol in superscript (i.e., X a).
Throughout this presentation, I will use commas to separate alleles. A slash mark (/) will be used to indicate heterozygosity for the trait proceeding it.
One method of symbolizing codominant alleles, which I will use in this presentation, is an upper case letter with various superscripts for the various mutations. The dominant Silver mutation is codominant with the wild type allele. This could be symbolized as M+ for the wild-type allele and MS for the Silver mutant allele.
If there are more than two alleles in a population (maximum of two per individual) for a particular gene locus, and an order of dominance exists, then the proper symbolization should be capital letter or wild type designation, if appropriate, for the most dominant gene, small case letter for the most recessive gene and small case with appropriate superscript for all the in between genes. For example: A > ax > ay > az > a
There are three methods of which I am aware for predicting the offspring of a particular breeding cross. These methods are the Punnett square, the Branching system and the FOIL method. Breeders should become familiar with each method and employ those methods that they feel most comfortable using.
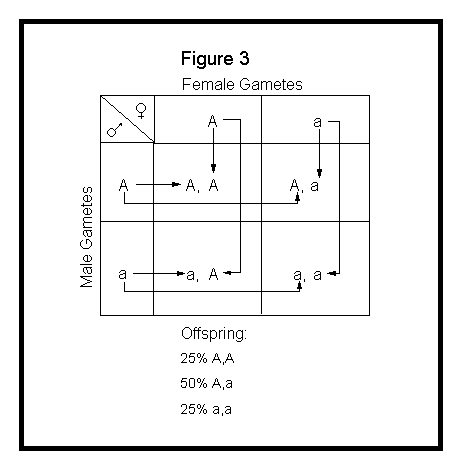
The Punnett square method simply involves lining up the gametes of one parent along the top of a checkerboard and the gametes of the other parent along the side, then showing the combinations within the cross-blocks. See Figure 3 below.
The Punnett square can be expanded to solve for more than a single trait (see figure 15) however, this method becomes very cumbersome when dealing with more than two different traits.
The Branching System method involves taking one set of parental genes and combining each of these genes with both genes of the other parental set. Figure 4 shows how the Branching system is applied to a genetic cross.
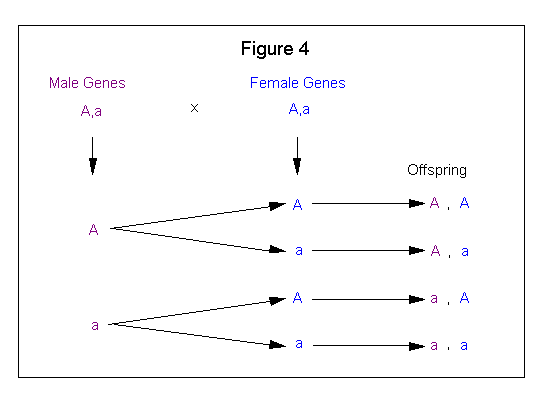
This method is very versatile (see Figure 17) and the method of choice for solving and displaying the more complex breeding crosses.
The FOIL method comes from the algebraic expression (a + b) (c + d) = ac + ad + bc + bd. This method, when applied to genetics, involves four steps as follows:
| 1. Combine the (F)-first of each pair. |
| 2. Combine the (O)-outer of each pair. |
| 3. Combine the (I)-inner of each pair. |
| 4. Combine the (L)-last of each pair. |
Figure 5 shows how the FOIL method is applied to a genetic cross.
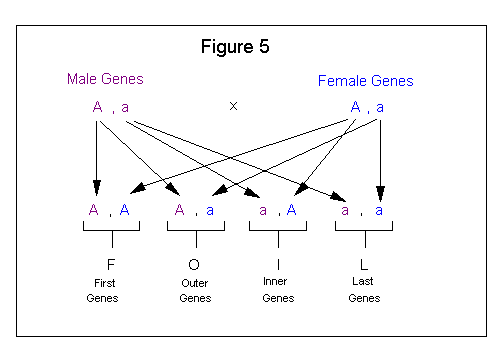
The FOIL method is easy to use but from a presentation standpoint does not lend itself very well to showing the sequence for how the outcome was derived. This method is pretty much done in your head; therefore, if you do not get easily confused you may want to use it. It can only be applied to a cross involving a single pair of alleles. If this method is used for crosses involving more than one trait then the individual gene pairs of the first cross need to be combined with the individual gene pairs of the second cross to show all possible outcomes. See Figure 6.

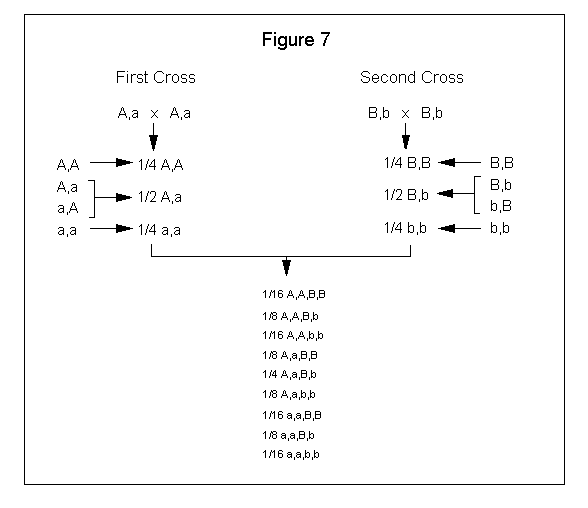
You can also derive the offspring probability by multiplying the first cross fraction with the second cross fraction as you combine the gene pairs. See Figure 7.